The microbiota in neonatal development is a complex bacterial ecosystem influenced by various factors such as the maternal microbiota, delivery, weight, antibiotics use, diet, etc. Antibiotics are administered for 72 hours per protocol because of the high probability of infection in the hospitalized neonates. The efficiency of such administration is in question because of the multiresistant profile of many of these bacteria and their unknown effects on the baby's microbiota. Infections with vertical maternal-neonatal transmission during pregnancy and childbirth are frequently caused by Streptococcus type B among others. One of the ways in which bacteria acquire these resistances to antibiotics is through horizontal gene transfer (HGT), usually from bacteriophages (phages) to bacteria. Phages encode and express auxiliary metabolic genes (AMGs) which are bacterial host genes that can impact host metabolic processes. Here, metagenomic data from a prospective mother-infant birth cohort in the Spanish-Mediterranean area has been used to look for correlations between phages and Streptococcus in the human gut microbiome. Comprehensive depurate databases for bacteriophages and Streptococcus have been developed. Then, a metagenomics pipeline to extract metagenome assembled genomes (MAGs) from Streptococcus genus and link them with phages has been developed. A total of 12 initial metagenomes (9 infants, 3 mothers) were used, obtaining 5 Streptococcus MAGs from 3 infant gut metagenomes that were selected for further analysis. AMGs of phages have been found in all of Streptococcus selected MAGs and encoded functionalities such as DNA metabolism (tyrosine recombinase XerC) or glycan biosynthesis and metabolism (lipoteichoic acid synthase) among others.
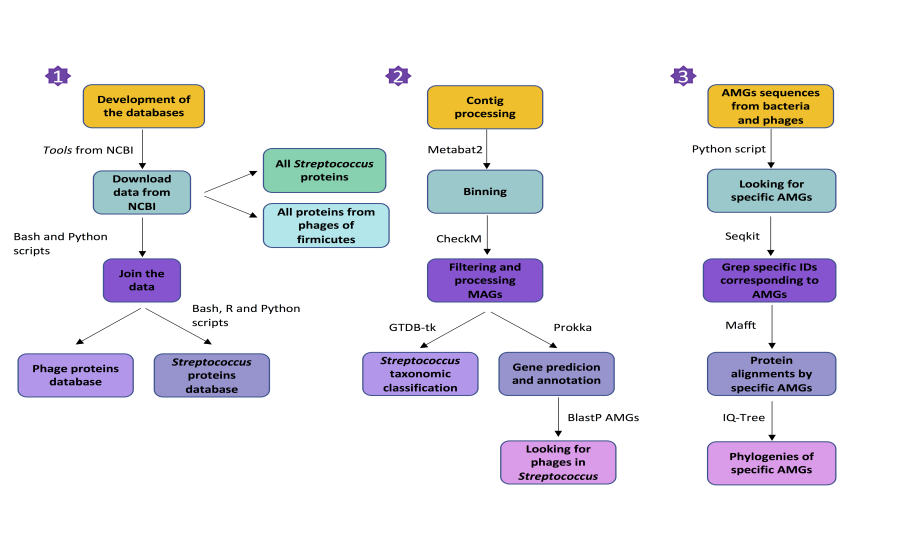
A. To create depurate “databases” of phages and Streptococcus.
• Looking for specific databases of phages and Streptococcus and downloading virus and bacterial data from NCBI.
• To develop Python scripts for creating the databases.
B. To implement a metagenomics pipeline for working from contigs and extracting Streptococcus MAGs.
• To compare metagenomics binning approaches and filter metagenome assembled genomes (MAGs).
• To assign selected MAGs to Streptococcus strains and annotate these MAGs.
• To associate phages with classified Streptococcus bins by looking for Auxiliary Metabolic Genes (AMGs) in both bacteria and viruses.
• Python3 is assumed to be installed (https://www.python.org/downloads/).
• samtools is assumed to be installed (http://www.htslib.org/)
• Metabat2 is assumed to be installed (https://bitbucket.org/berkeleylab/metabat/src/master/).
• CheckM is assumed to be installed (https://github.com/Ecogenomics/CheckM).
• BlastP is assumed to be installed (or DIAMOND).
• DIAMOND is assumed to be installed (https://github.com/bbuchfink/diamond).
• GTDB-tk is assumed to be installed (https://github.com/Ecogenomics/GTDBTk).
• Prokka is assumed to be installed (https://github.com/tseemann/prokka).
To develop databases, data from databases like NCBI can be downloaded with "Tools" or by the public repository by introducing key words such as phages. Annotations of phages in NCBI are not always correct, have a look on that.
With bash programming language, commands FASTA files containing the sequences can be prepared and joined in multiFASTA files for later use. The steps followed include: unzip all files, extract all FASTA files and join them, extract all the headers to understand the patter of FASTAs for creating the databases, and remove the duplicates with seqkit tool. With samtools tool (http://www.htslib.org/), databases can be also indexed. Finally, these files containing the sequences of genomic phages, genomic Streptococcus and proteins of phages have been compressed. To generate an output with the parameters of these databases, the fasta_csv_genomes.py and fasta_csv_proteins.py scripts have been used. This information is in the unix_commands_fasta.sh script.
Binning assembled sequences into individual groups which represent microbial genomes represents one of the most imporant steps in metagenomics, trying to recovery the complete information from all the population in one environment. Binning methods assume that the abundance of genes of the same species covary for the same taxon and/or that the contigs of the same bin have frequencies of use of k-mers (all possible combinations of nucleotides of length k that are contained in a similar sequence) (Dröge, Schönhuth, and McHardy 2017).
Depth coverage files, which calculate the average number of reads aligned to an individual base, has to be previously generated from FASTQ raw files by using Samtools and Bowtie2 tools.
This tool uses nucleotide composition information and source strain abundance to perform binning (Kang et al. 2019).
jgi_summarize_bam_contig_depths --outputDepth depth.txt *.bam
Then, we Run Metabat2 with the assembled FASTA files and the .txt depth coverage files.
Introduce:
-i: input in FASTA format
-a: depth.txt files
-o: generated output with binned sequences
metabat2 -i assembly.fasta -a depth.txt -o bins_dir/bin
After binning, a next step for filtering and preprocessing best-quality MAGs is necessary. CheckM tool is an automated method which assesses the quality of the virtual genome using a broader set of marker genes specific to the position of a genome within a reference genome tree and information about the collocation of these genes (Parks et al. 2015). This tool provides accurate estimates of genome completeness and contamination which helps to identify errors in metagenomes (Parks et al. 2015). Completeness evaluate how complete is a genome by presence/absence of unique marker genes, while contamination evaluates if genes appear more than once as two copies are usually lethal. By checking for domain-specific markers, we can also assess if our genomes have the recquired quality (Parks et al. 2015).
checkm taxonomy_wf -f outputname.txt --tab_table -t threads -x fa taxon binfolder outputfolder
Previously filtered, selected and taxonomically classified genomes by Streptococcus genus can be annotated with Prokka software (https://github.com/tseemann/prokka). This command line software annotates bacterial, archaeal and viral genomes quickly and produces output files for further analysis (Seemann 2014).
bins_dir: Output folder [auto] (default '')
--outdir: directory for output files
--cpus: Number of CPUs to use [0=all] (default '8')
--centre X: Sequencing centre ID. (default '')
-compliant: Force Genbank/ENA/DDJB compliance: --genes --mincontiglen 200 --centre XXX (default OFF)
#working
ls *.fa | parallel --verbose "prokka {} --prefix {.}_out"
In order to assign the previously filtered bins by CheckM to Streptococcus genus, Genome Taxonomy Database Toolkit (GTDB-tk) has been used (gtdb.ecogenomic.org). This tool provides an automated taxonomic classification of both bacterial and archaeal genomes by placing them into domain-specific, concatenated protein reference trees (Chaumeil et al. 2020).
Introduce:
classify_wf: The classify workflow consists of three steps: identify, align, and classify. This information can be found in (https://ecogenomics.github.io/GTDBTk/commands/classify_wf.html).
--genome_dir: directory containing genome files in FASTA format
--out_dir: directory for output files
--cpus: number of CPUs to use
-x: end of FASTA file
gtdbtk classify_wf --genome_dir dir-name --out_dir output-dir-name --cpus 20 -x fn
DIAMOND is a sequence aligner for protein and translated DNA searches, designed for high performance analysis of big sequence data which pairwase alignment of proteins and translated DNA (https://github.com/bbuchfink/diamond).
First, we create a database with DIAMOND
--in: input file
--db: name of the database
diamond makedb --in dir-name/multifasta_name.faa --db name_database-db
Then, we run BLASTp with the previous selected bins from Streptococcus against this database.
--more-sensitive: sensitivity of the tool
--matrix BLOSUM45: more distantly related alignments
--threads: Specifies the number of files which can be processed simultaneously
--db dir: database directory
--outfmt: a tabular text-based format produced by both BLAST+ output format 6
--query: to take the input files
--query-cover: number that describes how much of the query sequence is covered by the target sequence
--subject-cover:
--evalue: expect value. "E-value can be used as a first quality filter for the BLAST search result, to obtain only results equal to or better than the number given by the -evalue option. Blast results are sorted by E-value by default (best hit in first line)(https://www.metagenomics.wiki/tools/blast/evalue).
--out: name of the output
for i in *.faa; do diamond blastp --more-sensitive --matrix BLOSUM45 --threads 32 --db dir-name/name_database-db.dmnd --outfmt 6 --query $i --id 30 --query-cover 50 --subject-cover 50 --evalue 0.00001 -c1 --out $i-vs-output_name.blastp; done
Finally, the information from headers can be extracted from .faa annotation files of the desired bins from both AMGs of Streptococcus bins and bacteriophages from the database. With a script in R, we join these data by ID identification and we generate 2 new columns in an Excel (.*xlsx) format or (.csv) format. This will create a Microsoft Excel table with all the results.
With these tables, heatmaps can be created with ggplot2 in R heatmaps_AMGs.Rmd.
For visualizing the information from AMGs BlastKOALA has been also used: https://www.kegg.jp/blastkoala/
To create the phylogenies of specific families of proteins (i.e., tetracycline resistance, macrolide resistances, etc) we should download all the proteins of the bacterial genus we are interested (i.e.; Streptococcus) and concatenate them. Maybe we could also download just specific proteins of specific families (i.e.; tetracycline etc).
Then, we run BlastP of all Streptococcus proteins against the phages database previously built. The following script correponds to the first created database: phage_proteins-db.dmnd which contains all the proteins of phages of Firmicutes from NCBI:
for i in *.faa; do diamond blastp --more-sensitive --matrix BLOSUM45 --threads 32 --db /home/elena/phage_proteins-db.dmnd --outfmt 6 --query $i --id 30 --query-cover 50 --subject-cover 50 --evalue 0.00001 -c1 --out $i-vs-phages.blastp; done
Then, we take the second column from blastp results and create a new txt file with IDs to extract all the sequences for the alignment:
awk ' { F = FILENAME ".txt"; print $2 > F } ' *.blastp
Then, we take all the .txt files with IDs of interest and we run them into a database of proteins in FASTA format. We create a new FASTA file containing these sequences and we use a for loop to do it with all the possible .txt files and functionalities.
for i in *.txt; do seqkit grep -f $i /home/elena/databases/myphagesproteinsdb.faa -o $i-phages.faa; done
TAKE AMGs from BACTERIA: Print first column of ID (corresponding to bacteria)
awk '{print $1}' mystreptococcus_proteinsdb.faa-vs-phages.blastp > streptococcus_blastp_db_ID1.txt
Grep a specific file with .txt IDs corresponding to bacteria AMGs IDs and take the sequences corresponding to them:
seqkit grep -f streptococcus_blastp_db_ID1.txt mystreptococcus_proteinsdb.faa > selected_streptococcus_proteinsdb-bacteria.faa
Grep a specific file with .txt IDs corresponding to phages AMGs IDs and take the sequences corresponding to them:
seqkit grep -f streptococcus_blastp_db.txt PHAGESDB.faa > phagesdb-phages.faa
Focus on the following functionalities: INTEGRASE, MACROLIDE, TETRACYCLINE DNA-binding, integrase, recombinase, helicase, repressor others related to antibiotic resistance such as --> macrolide, tetracycline
To get FASTA sequences of specific functionalities we can run the script AMG_functions.sh and then obtain a multi FASTA from the specific functionality we were looking at.
To look for specific AMGs encoding for specific functionalities such as antibiotic resistance, HMM profiles could be built in order to find them correcly.
Profile HMMs are probabilistic models that encapsulate the evolutionary changes that have occurred in a set of related sequences (i.e. a multiple sequence alignment). To do so, they capture position-specific information about how conserved each amino acid is in each column of the alignment.
We use hhmbuild and hmmsearch in order to build the HMM profiles and then search them in our sequences.
hmmbuild tetO.hmm tetO.aln
hmmsearch tetO.hmm X.faa
Continue with alignment with mafft for different functionalities:
mafft [arguments] input > output
We create a IQ-TREE for generating phylogenies and with iTOOL we visualize trees and then use vector format to edit the phylogenies:
iqtree -s seqs.aln -pre output-name -bb 1000 -nt AUTO -alrt 1000
First example of phylogenies:
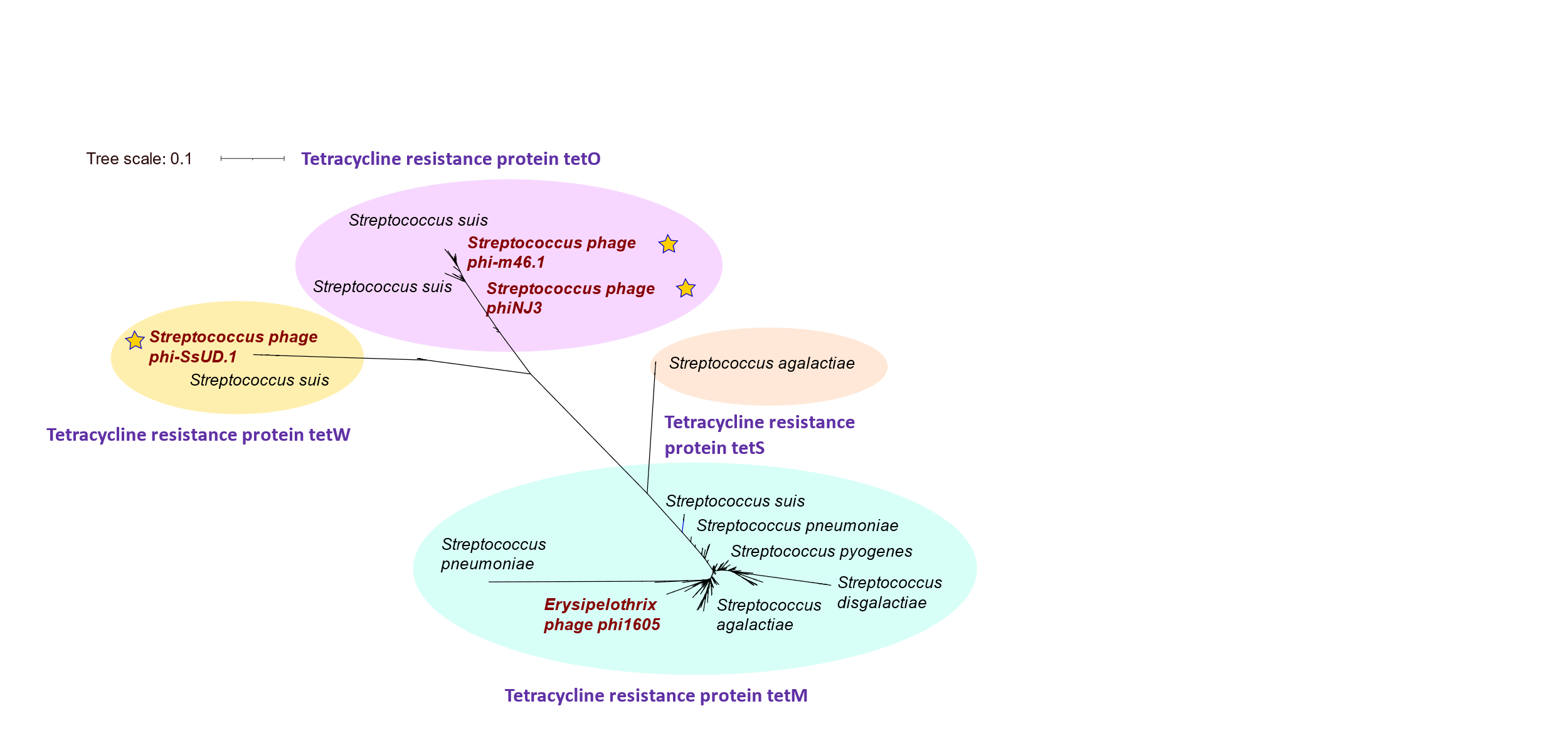
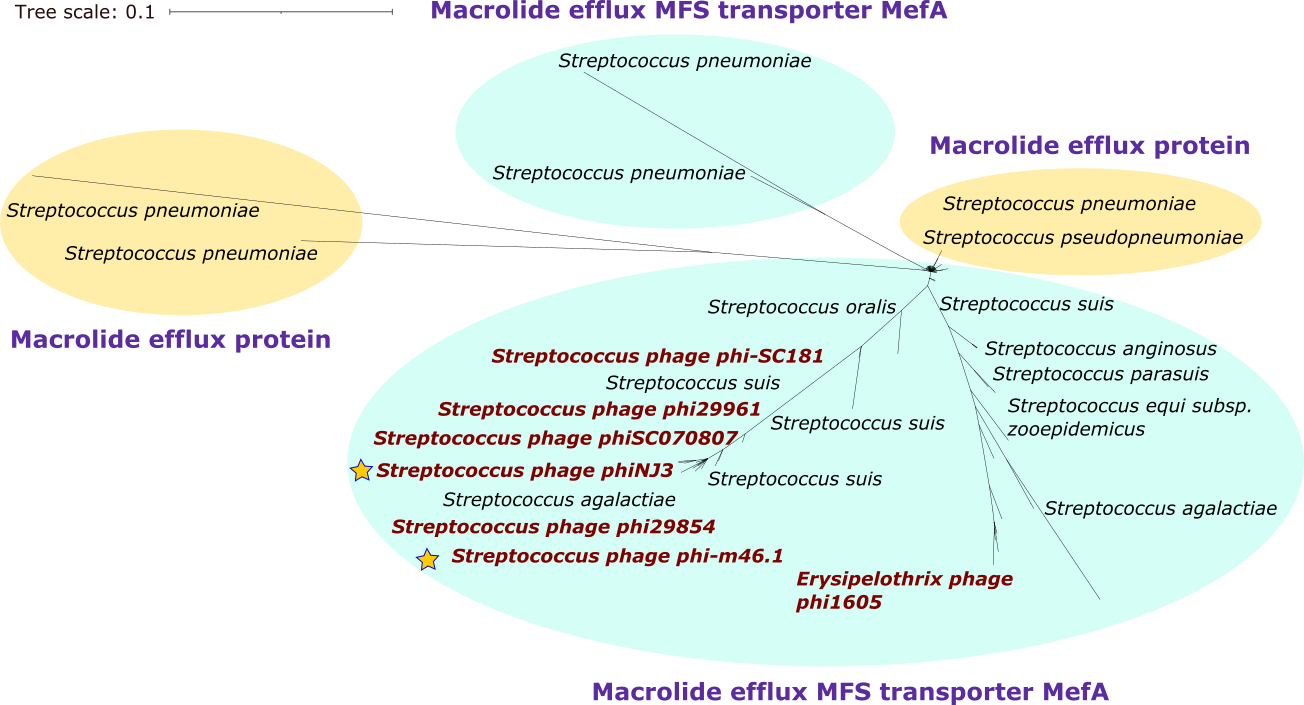
Together with the increased presence of antibiotic resistance in bacterial pathogens, bacterial viruses have been studied as possible therapeutic and biocontrol agents involving new people in bacteriophage research (Turner et al. 2021). For this reason, it is crucial to accurately annotate genomes. However, there are errors in assemblies which can lead to mistakes in annotation in databases such as NCBI. There are also proteins that belong to same categories and are thus missannotated, even if they have a different function. By manually searching the obtained proteins with a double check with BLASTp, some errors in bacteriophages annotations were found. Some of the hits found in the database of phages corresponded to bacterial contigs instead of phages. Thus, a rigorous sequence analysis should always be carried out. Thus, if there is a special interest in a community of bacteriophages, the database could be reduced and carefully studied previously.
Check these things with VIBRANT and compare with the AMGs pipeline results which are in the koch server in:
• Stored in: /home/elena/Pipeline_MAMI_Data
• Tetracycline resistance genes: /home/elena/Pipeline_MAMI_Data/strepto/prokka_annotation/all_faa_files/TETRACYCLINE_Streptococcus/all_faa_files/TETRACYCLINE_final_results
Chaumeil, Pierre-Alain, Aaron J Mussig, Philip Hugenholtz, and Donovan H Parks. 2020. “GTDB-Tk: A Toolkit to Classify Genomes with the Genome Taxonomy Database.” Oxford University Press.
Dröge, Johannes, Alexander Schönhuth, and Alice C McHardy. 2017. “A Probabilistic Model to Recover Individual Genomes from Metagenomes.” PeerJ Computer Science 3: e117.
Kang, Dongwan D, Feng Li, Edward Kirton, Ashleigh Thomas, Rob Egan, Hong An, and Zhong Wang. 2019. “MetaBAT 2: An Adaptive Binning Algorithm for Robust and Efficient Genome Reconstruction from Metagenome Assemblies.” PeerJ 7: e7359.
Parks, Donovan H, Michael Imelfort, Connor T Skennerton, Philip Hugenholtz, and Gene W Tyson. 2015. “CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes.” Genome Research 25 (7): 1043–55.
Seemann, Torsten. 2014. “Prokka: Rapid Prokaryotic Genome Annotation.” Bioinformatics 30 (14): 2068–69.
Turner, Dann, Evelien M Adriaenssens, Igor Tolstoy, and Andrew M Kropinski. 2021. “Phage Annotation Guide: Guidelines for Assembly and High-Quality Annotation.” PHAGE 2 (4): 170–82.